The Magrathea Manual: Coordinated Agent Modelling By Example
Contents
- 1 Introduction
- 2 Availability
- 3 Acknowledgements
- 4 Copyright
- 5 Introduction to Coordinated Agent Modelling By Example
- 6 Molecules and Coordinators
- 6.1 Coordinators act on molecules
- 6.2 Coordinators may be used to define complexes
- 6.3 Coordinator members have stoichiometry
- 6.4 Coordinators may be used to define compartments
- 6.5 Coordinators may be used to define membranes
- 6.6 Coordinators may be used to describe a molecule’s context
- 6.7 Coordinators may be used to organize the molecules in a simulation
- 7 Present implementation
- 8 Examples
- 8.1 Example 1. Point coordinator representing an interaction
- 8.2 Example 2. Point coordinator representing a complex
- 8.3 Example 3. Point coordinator representing a compartment.
- 8.4 Example 4. Plane coordinator representing a membrane section.
- 8.5 Example 5. Line coordinator representing a microtubule.
- 8.6 Example 6. Coordinators and rules.
Introduction
Magrathea is a prototype implementation of the Coordinated Agent Modelling (CAM) Method for representing biological pathways.
This prototype may be used to generate rule-based animated movies representing biological pathways. While it is hoped that this prototype demonstrates the underlying principals of CAM and that it is useful as an exploratory or narrative tool, no claims are made as to the utility or correctness of models generated using this method.
Availability
The magrathea.tz prototype script can be made available to interested collabortors. A paper is being prepared describing this work. These manual pages are posted for the benfit of collaborators and interested readers.
Comments, feedback and requests for the magrathea.tz prototype are welcome and may be sent to ian.donaldson@biotek.uio.no.
Acknowledgements
The Magrathea prototype and this documentation were developed by Ian Donaldson and Antonio Mora at the Biotechnology Centre of Oslo, University of Oslo.
The Magrathea prototype is dependent on the breve autonomous agent toolkit written by Jon Klein and available at http://www.spiderland .org.
Copyright
The material on this site is Copyright 2009, Ian Donaldson.
Introduction to Coordinated Agent Modelling By Example
This section outlines some of the defining features of Coordinated Agent Modelling (CAM). A more detailed and formal description of the implementation of CAM in the Magrathea prototype program is given in the second part of The Magrathea Manual: Building Coordinated Agent Models. This section describes the construction of model files for CAM models.
Coordinated Agent Modelling considers that animation of pathways and simulation of pathways belong to the same continuum. The Coordinator is a device that is used to tie animation and simulation to this same continuum. The following sections beginning with Molecules and Coordinators briefly review the ideas of CAM. The sections beginning with Present Implementation walks the reader through an overview of the prototype algorithm. Finally, the sections beginning Examples covers the ideas of CAM using a series of examples.
Molecules and Coordinators
Coordinated agent modeling is a method used to animate representations of biological pathways. A coordinated agent model is created by defining a set of molecule types and coordinator types.
In brief, molecule type descriptions define the properties of the types of molecules in the model. Coordinator type descriptions define the types of potential relationships that may occur between these different types of molecules. These relationships include physical interactions and any molecular transformations that may occur as a result of these interactions. In addition to describing the types of molecules and coordinators, the model may also specify the number and locations of the molecules and coordinators that initially populate the simulation.
A model is initially populated with molecules distributed in some three-dimensional space. Coordinators are brought into existence when two molecules move within one another’s neighborhoods (say, for instance, they collide) and those two molecules are both potential members of some coordinator (such as a potential interaction). When the coordinator is created, it is assigned a position in three-dimensional space that is midway between its member molecules.
Coordinators act on molecules
Coordinators act on molecules. For instance, a coordinator defining an interaction between two molecules will adjust the physical locations of these two molecules at each time step of the simulation so they remain adjacent to one another without overlapping. Each coordinator maintains a list of the molecules that it is currently acting on (its member molecules). A coordinator’s members may change over the course of the simulation. Conversely, each molecule maintains a list of the coordinators that are acting on it.
A coordinator acts on its member molecules according to a set of rules predefined in the coordinator type description. These rules may cause the member molecule to undergo state changes or translocations (across a compartment boundary). The execution of these events is dependent on a set of conditions that must be met. Conditions may include the number, types and states of the other member molecules of the coordinator. Coordinator rules may also be used to determine when molecules of some type may either join or leave the coordinator.
Coordinators may be used to define complexes
A coordinator with two member molecules is used to describe an interaction between those two molecules along with the resulting molecular transformations that take place as a result of the interaction.
A coordinator may also be used to define an interaction between three or more molecules in a complex without defining the individual interactions between the members of the complex. This may be useful in cases where the interactions are either unknown or of no importance to the model. In cases where a coordinator defines a complex, the member molecules are constrained in a region of space defined by the surface of a sphere with the coordinator at the centre. The surface area of the sphere is equivalent to the sum of the cross-sectional areas of the member molecules. Each molecule is held to the surface of this sphere using a spring function defined by the positions of the coordinator and the molecule.
Coordinator members have stoichiometry
Coordinators have potential members defined by a molecule type and some maximal stoichiometry for that molecule type. A coordinator is created once two or more potential molecules encounter one another in their respective neighborhoods. Additional members may join this coordinator in subsequent time-steps if the maximal stoichiometry for that molecule type has not been met. The addition of molecules to a coordinator may be unordered or (optionally) ordered by a set of addition rules specified by the coordinator. Similarly, rules may be used to specify the order in which molecules leave a coordinator depending on the presence, stoichiometry and state of any of the member molecules.
Coordinators may be used to define compartments
A coordinator may be used to describe a compartment. The representation is similar to that of a complex. Again, the coordinator is positioned midway between member molecules. The coordinator then moves (and holds) these molecules to a position on the surface of an imaginary sphere whose surface area is equivalent to the sum of the cross-sectional areas of each of the members radii. The resulting structure appears as a spherical compartment whose surface is covered by the member molecules in a closely packed configuration. Optionally, a set of “marker” molecules may be assigned as members of the coordinator that have an “effective radius” that is larger than their apparent (visualized) radius. This allows for the demarcation of large compartments without having to specify molecules that cover the entire surface.
Molecules may be described as potential inhabitants of compartments. This acts to restrict molecules from entering compartments where they do not belong and to constrain molecules within compartments where they do belong. Molecules may be moved into and out of compartments by interacting with surface molecules (members of the coordinator representing a compartment) that act as transporters. This interaction and subsequent translocation is encoded by a set of coordinator rules. This translocation may be dependent on the state of the transporter molecule and/or the state of the molecule to be transported.
Coordinators may be used to define membranes
Coordinators representing interactions, complexes and compartments all use a point location around which member molecules are organized. Coordinators have two other geometric forms which allow for the representation of planar and linear macromolecular structures.
Planar coordinators may be used to represent membranes. Member molecules of planar coordinators are constrained to movement in the surface of the plane using a spring function defined by the positions of the coordinator and the molecule. Non-member molecules are restricted from passing through a planar coordinator although they may be translocated across the plane by a method analogous to that described for compartment transporters.
Linear molecules may be used to represent structures such as microtubules. Member molecules are constrained to movement along a linear path defined by a line with some beginning and end point in 3D space.
Coordinators may be used to describe a molecule’s context
A molecules function is largely dependent upon its context. A molecule’s context may be thought as the sum of all the sub-contexts in which that molecule is directly a member.
For example, a membrane-bound receptor (r) may bind to some extra-cellular ligand (l) to form an rl complex. The formation of the complex initiates some intracellular signalling event. In this example, r may be thought of as having two potential contexts. The first context is that of the membrane (M). r being in the context of M has implications on its physical location and movement as well as the molecules that it is most likely to meet with (other molecules with context M). The second context is that of a specific interaction (I) between r and l. r being in the context of I has implications on its physical location (it will of course co-localize with l) and on the probability of events associated with this context (for example, the transformation of r to some alternative “active” state that will allow r to participate in some new context; say some downstream event in the signalling pathway).
In this example, the receptor (r) has two sub-contexts; an interaction (I) and a membrane (M). Both of these contexts may be represented by coordinators in a CAM model (see above). The ability of R to simultaneously participate in two sub-contexts is described by assigning two “channels” to R. A channel represents the potential of a molecule to be coordinated by some coordinator. In this case, channel A conveys effects on R encoded by coordinator I and channel B conveys effects encoded by coordinator M. When molecule R joins a coordinator, it does so by assigning that coordinator to a channel and that channel is then said to be occupied. Each channel may be unoccupied or occupied by a single coordinator from a list of potential coordinator types for that channel. This “channel” device allows a molecule to simultaneously participate in (be effected by or informed by) multiple sub-contexts. A channel may correspond to a physical binding site though this does not hold in all cases (such as for the channel mediating the membrane (M) context).
The above example may be extended to include an effector molecule (e) that binds to the cytosolic portion of the receptor (r). This interaction is represented by a third coordinator (S). S may encode a rule that monitors the states of the member molecules at each time-step. If r is in some activated state and if e is present in some inactive state, then the rule specifies an event whereby some feature of e changes state and leaves the coordinator. This example demonstrates how a model may propagate a state-change (signal) from one molecule to another (l to r to e) via coordinators (I and S) even though these two molecules (l and e) do not directly share the same context.
Coordinators may be used to organize the molecules in a simulation
Coordinators are brought into existence once two molecules occur within one another’s neighborhoods and those two molecules can both potentially belong to the same type of coordinator. A molecule’s neighborhood is defined as a sphere with some radius that has the same centre as the molecule. If two molecules each have neighborhoods with radii equivalent to their own radii, a potential coordinator may be created when these two molecules collide.
The neighborhood size may be specified to be larger than the actual molecule. At each time step of the simulation, the molecule will check its neighborhood for the presence of other molecules with which it could create a coordinator. This device allows for a coordinator defining an interaction to be created before the two molecules actually collide. The coordinator defining the interaction will then act to move these two molecules together and maintain their association.
Additionally, the neighborhood size of a molecule may be set to increase at each time-step of the simulation if the molecule has unoccupied channels. This device is called “dynamic neighborhood sizing” and has the effect of allowing each molecule to scan the simulation until it finds its closest potential coordinator co-member. Dynamic neighborhood sizing has the effect of organizing the molecules of the simulation such that they become animated by a set of predefined rules encoded by coordinators.
As molecules become assembled with coordinators, they may change state, be destroyed, be translocated, leave existing coordinators, join existing coordinators or create new coordinators. Since coordinators are dynamically formed and destroyed according to the local presence of molecules and their individual states, the components of the model are constantly organized and re-organized throughout the time course of the animation.
If the neighborhood size of molecules is set to zero, then coordinators are formed when molecules physically collide. If dynamic neighborhood sizing is used, molecules will be organized by coordinators before they collide. This device then ties simulation (neighborhood size = 0) and animation (dynamic neighborhood sizing) to the same continuum.
Next we introduce the present implementation of the CAM method and demonstrate how it may be used to build such animated narratives of biological pathways.
Present implementation
The CAM algorithm has been prototyped using the Breve autonomous agent programming toolkit using the object-oriented steve scripting language (http://www.spiderland.org). The prototype script is named magrathea.tz and is available from ian.donaldson@biotek.uio.no.
Major classes employed by the Magrathea prototype
Magrathea has four major classes. moleculeType encodes properties and methods that are common to all molecules of a given type. This class essentially serves to hold redundant information and is employed by method calls that return information about the molecule type. molecule encodes properties and methods that are specific to an instance of some molecule type. Similarly, coordinatorType and coordinator encode properties and methods specific to coordinators. A model is loaded from a text file that specifies moleculeType and coordinatorType parameters as well as a description of the initial instantiations of molecules and coordinators belonging to these types.
Overview of the algorithm
The figure below shows the core set of functions that are carried out during each iteration of the simulation. Below we describe three of these functions in greater detail. assign-to-coordinators is used to assign molecules to coordinators (either to existing coordinators or to the creation of new coordinators) in a dynamic manner according to a molecule’s local context. move is used to move molecules according to their membership in these coordinators. process-rules is a coordinator function that effects events on member molecules based on a set of rules. Events include state-changes, transformations and translocations of molecules among other things. Rules encode a set of preconditions that must be met before some event can be executed with some probability encoded by a set of parameters.
+to iterate:
self update-neighbors.
molecules = all Molecules.
coordinators = all Coordinators.
foreach item in molecules: {
item assign-to-coordinators.
item process-mol-rules.
}
foreach item in coordinators: {
item reposition.
item prune-redundant-coordinators.
}
molecules = all Molecules.
coordinators = all Coordinators.
foreach item in coordinators: {
item process-rules.
item remove-unassembled-molecules.
item prune-redundant-coordinators.
molecules = all Molecules.
}
foreach item in molecules: {
item move.
}
//
Movie and camera movement functions
//
super iterate.
assign-to-coordinators
Each molecule type has a list of coordinator types in which it may potentially participate. In turn, each coordinator type contains a) a list of molecules types that may join the coordinator b) the maximal stoichiometry of each molecule type c) an optional set of assembly rules that restrict the addition of molecules to the coordinator as a function of any present or potential members of the coordinator and their states. As an example, a coordinator may describe two molecules that may interact. At each time step in the simulation, each molecule will examine its local neighborhood for the presence of other molecules with which it may form a coordinator. The local neighborhood is defined as a sphere centered on the molecule and with a diameter of some “neighborhood size”. If the molecule finds at least one other molecule in its neighborhood with which it may form a coordinator (as defined by its coordinator types and the parameters of the coordinator) then that coordinator is formed and both molecules are assigned as members to that coordinator. In the case where multiple coordinators are possible, the coordinator which is able to involve the greatest number of molecules is chosen. Typically, a simulation will begin with no coordinators and their creation is dynamic. Once coordinators exist, free molecules (with no coordinator assignments) may join an existing coordinator in the neighborhood. Conversely, a molecule assigned to a coordinator may leave that coordinator to join an alternative coordinator within its neighborhood (competition between coordinators is described further in the additional materials).
When a molecule is assigned to a coordinator, that coordinator is said to “occupy” a “channel” on the molecule. Molecules have a finite set of channels (typically 1 or 2) that can be roughly thought of as potential binding sites. More correctly, each channel represents the ability of the molecule to “be informed by” or “to listen to” or “participate in” some coordinator. Each of a molecule’s channels may be occupied by only one coordinator at a time.
If any one of a molecule’s channels remains unoccupied by the end of some iteration, the molecule’s neighbourhood size may be optionally increased by some step size in the next iteration. This option is referred to as dynamic neighborhood sizing (DNS) and serves to allow the molecule to “find” other coordinator co-members without having to directly encounter them by diffusion. DNS also ensures that a molecule forms or joins coordinators based on the closest possibilities. Typically, the neighborhood size starts off as equivalent to the radius of the molecule. If we were to disallow DNS for two example molecules, they would simply move randomly and the coordinator (say an interaction) would not be created until the two molecules physically collided with one another (as determined by their location and radii). Once all of a molecule’s channels are occupied, its neighborhood size is reset to its starting size. The neighborhood size is also reset if the molecule changes state or if a potential coordinator co-member (anywhere in the simulation) changes state.
move
This function is used to calculate and apply a new velocity to each molecule at each iteration. The new velocity of the molecule is calculated using the sum of three “forces”. The three forces include:
1) a random force applied to mimick brownian motion. 2) a coordinator force that organizes or constrains the molecule according to all coordinators of which it is a member. This coordinating force has been described above. 3) a spacing force, which serves to mimick collisions and prevent molecules from moving through one another. Collisions between molecules are never explicitly detected; instead, each molecule examines its six closest neighbors for any overlap. If an overlap is found, a repulsive force is applied that is proportional to the degree of overlap. Collisions between molecules and coordinators that represent compartments are handled by a separate function collide. This function serves to exclude molecules from compartments (to which they do not belong) or to prevent molecules from leaving a compartment (to which they do belong).
The “forces” described above are not intended to represent real molecular motion but serve to distribute, organize and constrain components of the model in space respective to one another.
process-rules
Coordinators hold information about events that may involve member molecules. By encoding this information within the coordinator (instead of the molecules), events for molecules occur according to their context (coordinator membership). This information is encoded by a set of rules. Each rule is composed of three parts: 1) a set of conditions that must be met before further consideration of the rule 2) a set of parameters that is used to probabilistically determine whether events are to be executed if the pre-conditions are met and finally 3) a set of events that are then applied to the condition meeting molecules. Competing rules Molecule rules
conditions: A rule must have a set of one or more conditions. Each condition specifies a molecule type that must be present in some stoichiometry and have (or not have) some specified set of feature-state values. Molecules may have an integer number of features. Each feature may exist in one of a number of integer states. A molecule’s feature-states may be specified at the beginning of the simulation and may be altered by an event (see below). Further, conditions may specify whether the molecule is UNASSEMBLED or ASSEMBLED with the present coordinator (see below under ordered assembly). Each condition is used to assign a tag to a set of condition meeting molecules (if found). This tag is used in the event description portion of the rule. parameters: Each rule has a parameter list consisting of key value pairs. Typically, one parameter is provided that represents the probability that the rule’s events will be executed given that the conditions are met. events: Each rule must have a set of one or more events. Events include
1) destruction of a molecule, 2) creation of a molecule at the position of some molecule, 3) changing a feature-state value of a molecule, 4) translocating a molecule to a specified compartment or past a membrane, 5) allow for a molecule to begin assembly with a coordinator, 6) allow for a molecule to leave a coordinator, or 7) changing the apparent colour of a molecule. Event descriptions include a tag that points to a condition meeting molecule(s) to which the event is to apply.
Ordered assembly
Molecules that are assigned as members of a coordinator may exist in one of three states with respect to the coordinator: UNASSEMBLED, ASSEMBLING or ASSEMBLED. Once a molecule is assigned to a coordinator (by assign-to-coordinators), it is considered to be UNASSEMBLED. A molecule’s assembly with a coordinator type is either unconditional or it is dependent on a set of conditions specified by some coordinator rule. If a molecule’s assembly is unconditional, the molecule immediately changes from UNASSEMBLED to ASSEMBLING once it has been assigned to the coordinator. Once a molecule is ASSEMBLING, it is subject to coordinator forces which will move the molecule towards the coordinator according to the geometry of the coordinator (either point, linear or planar) as described above. Alternatively, if a molecule’s assembly with a coordinator is conditional, the coordinator will execute an event called begin-assembly if conditions are met to initiate this assembly. These conditions may require that the molecule and other assigned but UNASSEMBLED molecules first be present with some specified feature-state values before assembly can begin. This scheme allows an order of assembly to be encoded. Assigned molecules which fail to become ASSEMBLING after considering these rules in process-rules are removed by remove-unassembled-molecules. In practice, a look ahead function (in assign-to-coordinators) is used when assigning molecules to coordinators (or when creating new coordinators) such that molecules will not be assigned to a coordinator unless conditions can be met to allow their assembly. This serves to limit the number of futile assignments and maintain molecules that cannot be assembled in an unoccupied state.
Examples
In this section, we describe a number of examples that demonstrate the capabilities of the prototype implementation of the CAM method.
Examples are described using movies and explanatory text that notes how molecules and coordinators are defined to achieve the described model capabilities. Model files are available for each example that detail the exact implementation of these methods. These model files are available at Example model files for Magrathea. These files may be directly copied and pasted into a file called “model.txt” that exists in the same directory as the magrathea.tz prototype script.
The user is referred to the Magrathea Manual: Building Coordinated Agent Models for a more detailed description of the syntax used in these model files. These model files are working examples; see the sections on Installation and Use for details.
Example 1. Point coordinator representing an interaction
  |
This is a simple example using two molecules labeled a and b. Each molecule is represented by a sphere with a radius of 1 unit and each has dynamic neighborhood sizing turned on. At each time step, the two molecules increase their neighborhood size by 1 until each is found within the other’s neighborhood. At this point, a coordinator (represented by a cube) is created and moved to a position midway between the two molecules. At each time-step thereafter, the coordinator moves the two molecules into position on an imaginary sphere drawn around the coordinator and holds them in this position using a spring function.
Click on the picture to view the movie. Your web browser requires a plugin (like Quicktime) that is capable of viewing mpeg files. Click the back button in your browser to return here when you are done viewing the movie. See interaction1.txt model file at Example model files for Magrathea The blue sphere at the centre of this image is a marker used in all simulations to show the world centre with x and y axes marked. A floor is also shown in all simulations for the z=0 plane. If dynamic neighborhood sizing is turned off (set to 0), the two molecules will most likely not collide with one another and will continue in a random walk through the simulation. |
Example 2. Point coordinator representing a complex
  |
This is a simple example using three molecule types labeled a, b and c. There are two molecules of type c. Each molecule is represented by a sphere with a radius of 1 unit and each has dynamic neighborhood sizing turned on. At each time step, the molecules increase their neighborhood size by 1 until at least two find one another within the other’s neighborhood. At this point, a coordinator (represented by a cube) is created and moved to a position midway between the molecules. This coordinator represents a complex with stochiometry 1a, 1b, and 1c. Only 2 molecules (of the 4 present) are required to initiate the formation of the complex since the assembly has no specified order. The third may join this coordinator later.
At each time-step thereafter, the coordinator moves all three molecules into position on an imaginary sphere drawn around the coordinator and holds them in this position using a spring function. The fourth (extra) molecule c remains unassembled. Click on the picture to view the movie. Your web browser requires a plugin (like Quicktime) that is capable of viewing mpeg files. Click the back button in your browser to return here when you are done viewing the movie. See complex1.txt model file at Example model files for Magrathea
|
  |
A second model file (complex2.txt) specifies a coordinator with a stoichiometry of 1a, 1b, 2c so all four molecules are included in the final complex.
Click on the picture to view the movie. Your web browser requires a plugin (like Quicktime) that is capable of viewing mpeg files. Click the back button in your browser to return here when you are done viewing the movie. See complex2.txt model file at Example model files for Magrathea You may notice at the beginning of the complex2.mpeg movie that a coordinator first forms between the 2 c subunits and then this coordinator moves between all four subunits. The coordinator first forms between the two c subunits since they are closest to one another in the simulation. When subunits a and b detect either of these c subunits in their neighborhood, they first check for existing coordinators that they can join. When a and b join this existing coordinator, the coordinator moves to a position midway between all four subunits. |
Example 3. Point coordinator representing a compartment.
  |
This is a simple example of a coordinator used to represent a compartment.
Twenty instances of a single molecule type become members of this coordinator (i.e. they define its boundary). The 20 molecules are constrained to movement on the surface of an imaginary sphere whose surface area is equivalent to the sum of the cross-sectional areas of the 20 molecules. The coordinator representing a compartment must be a point coordinator (see geometricType). In this simulation it is represented by a translucent sphere that grows from an initial size to the size of the imaginary sphere as molecules are added to it. This size increase is dependent on the line isSizeDynamic 1 The sphere is used to handle collisions with other molecules that are not a part of the compartment coordinator (see below). Click on the above image to view the movie of the simulation. See compartment.1.txt model file at Example model files for Magrathea
|
  |
Molecules may also be described that have a larger effective radius than what is rendered. This allows for the compartment to be delimited by “marker molecules” without having to cover the entire surface of the compartment. An example is shown in compartment.2.txt model file. A single change was made to the above model file to achieve this. See the line
shape Sphere 1 2 Click on the above image to view the movie of the simulation. See See compartment.2.txt model file at Example model files for Magrathea |
Example 4. Plane coordinator representing a membrane section.
 
  |
This is a simple example in which 20 instances of one molecule type are all potential members of a plane coordinator. The molecules are moved to the surface of the plane where they can move randomly in that surface but they cannot move out of the surface.
Plane coordinators are generally created at the beginning of the simulation and will persist for the duration of the simulation (i.e. they are not dynamically created according to context ant they cannot be destroyed). Further, plane coordinators can be specified as non-mobile so the plane remains stationary during the simulation (otherwise, the plane would move to maintain a centre that is midway between the positions of the member molecules). Lastly, like point coordinators representing compartments, the plane coordinator can increase in size from its starting size as member molecules join. These coordinator characteristics are encoded by the lines isCreationDynamic 0 isMobile 0 isSizeDynamic 1 in the [coordinatorType] section of the model file. Click on either image to view the movie of the simulation. Both figures link to the same movie. See See membrane.1.txt model file at Example model files for Magrathea Finally, the orientation of the plane at the start of the simulation can be changed using the orientation line. This model file can be altered by uncommenting (removing the # symbol) in front of one of the lines beginning with #orientation to see this effect. An example is shown in the bottom figure. Both figures link to the same movie. |
|
| |
 
  |
This example adds non-membrane molecules to either side of the membrane to show that collisions are handled with the surface of the membrane.
Click on either image to view the movie of the simulation. Both figures link to the same movie. See See membrane.2.txt model file at Example model files for Magrathea Again, the orientation of the plane at the start of the simulation can be changed using the orientation line. This model file can be altered by uncommenting (removing the # symbol) in front of one or more of the lines beginning with #orientation to see this effect. An example is shown in the bottom figure. |
Example 5. Line coordinator representing a microtubule.
 
  |
This is a simple example in which 20 instances of one molecule type are all potential members of a line coordinator. The molecules are moved to the line where they can move randomly along that line.
Like the plane coordinator described in the example above, the line coordinator shown in this model is created at the beginning of the solution and persists throughout the simulation without changing position. Click on either image to view the movie of the simulation. Both figures link to the same movie. The figures show the line coordinator in two different starting orientations. See See microtubule.1.txt model file at Example model files for Magrathea The associated movie in the example directory demonstrates use of camera movements during the simulation. The model file also shows an example of populating a simulation with a set of molecules that are arranged in a line using the instantiate line in the model file: instantiate 20 random cylindrical volume 10 15 10 0 1 0 0 35 See the section on building model files for more details. |
Example 6. Coordinators and rules.
  |
This is a simple demonstration of the use of rules by coordinators.
This example shows a substrate (molecule 1) that undergoes a state-change (phosphorylation) upon interaction with a kinase (molecule 2). The state change is visualized by a colour change. A single coordinator type uses three rules to control 1) the assembly of unphosphorylated substrate with the kinase, 2) the phosphorylation (state change) of the substrate and 3) the disassembly of the kinase-substrate complex after phosphorylation. After the substrate is phosphorylated, it can no longer be considered as a potential member of this coordinator and so the two molecules drift apart.
|
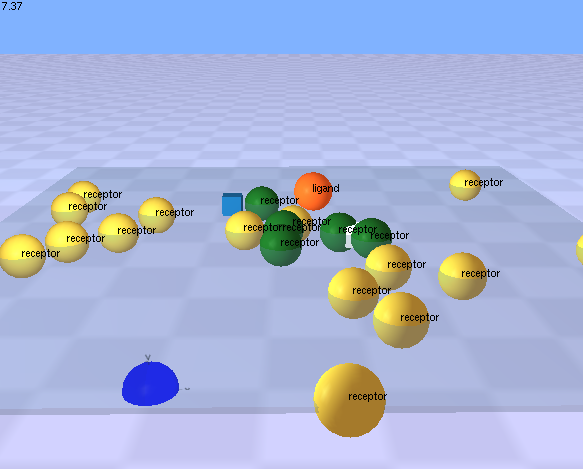  |
This demonstration shows a somewhat more complex use of rules to demonstrate the process of lateral signalling.
In this example, a ligand molecule binds to a membrane-bound receptor. On binding, the receptor changes state (becomes activated) as indicated by a colour change. The receptor in this new state also gains the ability to bind to and activate other membrane bound receptors. The overall effect is that a single ligand-receptor binding event initiates a lateral cascade of receptor activation events. Click on the picture to view the movie. Your web browser requires a plugin (like Quicktime) that is capable of viewing mpeg files. Click the back button in your browser to return here when you are done viewing the movie. See rules.1.txt model file at Example model files for Magrathea |